- Whats this?
- Principal Component Analysis (PCA)
- Spectral Clustering
- t-Distributed Stochastic Neighbor Embedding (t-SNE)
- Sources
Whats this?
It’s a document on PCA, Spectral Clustering and t-SNE, duh? All of these are used to make sense of the structure of data. More importantly, all of these are intricately related to eigendecomposition (Matrix Decompositions 1).
Why do I write this?
As always, I wanted to create an authoritative document I could refer to for later. Thought that I might as well make it public.
Principal Component Analysis (PCA)
What’s this used for? We use it to find a lower-dimensional linear subspace of a high-dimensional data space that captures the maximal amount of variance in the data.
Big dimensional data bad, lesser dimensions good:)
Perspectives
We can think about PCA from two viewpoints
- Maximizing the variance of projected data
- Minimizing reconstruction error
Huh? What’s variance? Covariance?
Let’s nail down expected value first.
The expected value of a random variable, which represents its theoretical mean or center of mass. For a continuous random variable 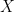
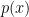
![\mathbb{E}[X] = \mu = \int_{-\infty}^{\infty} x p(x) dx](https://s0.wp.com/latex.php?latex=%5Cmathbb%7BE%7D%5BX%5D+%3D+%5Cmu+%3D+%5Cint_%7B-%5Cinfty%7D%5E%7B%5Cinfty%7D+x+p%28x%29+dx&bg=ffffff&fg=000&s=0&c=20201002)
So, as you can probably gauge, this is like the mean – describes the central tendency of a distribution. But, it does not tell us anything about its spread / dispersion. This is where we get variance!
The variance of a random variable 
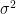
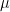
![\text{Var}(X) = \sigma^2 := \mathbb{E}[(X - \mu)^2]](https://s0.wp.com/latex.php?latex=%5Ctext%7BVar%7D%28X%29+%3D+%5Csigma%5E2+%3A%3D+%5Cmathbb%7BE%7D%5B%28X+-+%5Cmu%29%5E2%5D&bg=ffffff&fg=000&s=0&c=20201002)
The square root of the variance, 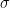
Takeaway – A low variance indicates that the values are tightly clustered around the mean, while a high variance indicates that they are spread out over a wider range.
Some math-y aspects of variance
The Raw-score Formula
The definition of variance can be expanded using the linearity of expectation.
![\text{Var}(X) = \mathbb{E}[(X^2 - 2\mu X + \mu^2)] = \mathbb{E}[X^2] - 2\mu\mathbb{E}[X] + \mu^2 = \mathbb{E}[X^2] - 2\mu^2 + \mu^2](https://s0.wp.com/latex.php?latex=%5Ctext%7BVar%7D%28X%29+%3D+%5Cmathbb%7BE%7D%5B%28X%5E2+-+2%5Cmu+X+%2B+%5Cmu%5E2%29%5D+%3D+%5Cmathbb%7BE%7D%5BX%5E2%5D+-+2%5Cmu%5Cmathbb%7BE%7D%5BX%5D+%2B+%5Cmu%5E2+%3D+%5Cmathbb%7BE%7D%5BX%5E2%5D+-+2%5Cmu%5E2+%2B+%5Cmu%5E2&bg=ffffff&fg=000&s=0&c=20201002)
![\text{Var}(X) = \mathbb{E}[X^2] - (\mathbb{E}[X])^2](https://s0.wp.com/latex.php?latex=%5Ctext%7BVar%7D%28X%29+%3D+%5Cmathbb%7BE%7D%5BX%5E2%5D+-+%28%5Cmathbb%7BE%7D%5BX%5D%29%5E2&bg=ffffff&fg=000&s=0&c=20201002)
This is more convenient for us. Why?
Empirally determining variance
In practice, we do not have the true distribution 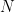


The empirical variance is the average of the squared deviations from this empirical mean:

Important note – The unbiased estimator uses a denominator of 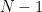
Huh? You ask again.
Why did you talk
First – an unbiased estimator is a formula that, on average, gives us the correct value for the property that we are trying to measure from a population.
The population bit is important – this is the actual disribution. What are have are samples!
When you calculate the variance of a sample, you don’t know the true mean (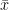
The sample mean (
for that particular sample.
Since the sample mean is always a “perfect fit” for the sample, the deviations from it are slightly smaller than the deviations would be from the true population mean! So, we divide by
Using a smaller denominator makes the final result slightly larger. This small increase perfectly corrects for the underestimation caused by using the sample mean, ensuring that, on average, the calculated variance matches the true population variance. This is why it’s called the unbiased estimator. (What’s this? Keep reading until a few more sections. Or hit ctrl+F and find biased-estimator-def)
Ok, but, why
(Feel free to skip this bit if you don’t care about Math. But, then, that’s why you are here, isn’t it?)
First, let’s define our terms:
is the sample mean.
- The sample variance estimator is
.
- The expectation operator,
, gives the long-run average of an estimator. For an estimator to be unbiased, its expectation must equal the true parameter, i.e.,
.
Now, the derivation!
Our goal is to compute the expected value of the sum of squared differences,
. We start by rewriting the term inside the sum:
Expanding the squared term gives:
Distributing the summation:
Now, let’s simplify the middle term,
, which equals
. Substituting this in:
Now we take the expectation of this entire expression:
By definition,
is the population variance
is the variance of the sample mean, which is
. Substituting these in:
See? Now remember that you can user either
since for very, very large
.
Covariance!
Variance describes how a single variable changes. Covariance extends this idea to describe how two variables change together.
The covariance between two random variables
with means
and
respectively, is the expected value of the product of their deviations from their means:

![= \sum\left[ (x_i - \mu)^2 - 2(x_i - \mu)(\bar{x} - \mu) + (\bar{x} - \mu)^2 \right]](https://s0.wp.com/latex.php?latex=%3D+%5Csum%5Cleft%5B+%28x_i+-+%5Cmu%29%5E2+-+2%28x_i+-+%5Cmu%29%28%5Cbar%7Bx%7D+-+%5Cmu%29+%2B+%28%5Cbar%7Bx%7D+-+%5Cmu%29%5E2+%5Cright%5D&bg=ffffff&fg=000&s=0&c=20201002)




![E\left[\sum(x_i - \bar{x})^2\right] = E\left[\sum(x_i - \mu)^2\right] - E\left[N(\bar{x} - \mu)^2\right]](https://s0.wp.com/latex.php?latex=E%5Cleft%5B%5Csum%28x_i+-+%5Cbar%7Bx%7D%29%5E2%5Cright%5D+%3D+E%5Cleft%5B%5Csum%28x_i+-+%5Cmu%29%5E2%5Cright%5D+-+E%5Cleft%5BN%28%5Cbar%7Bx%7D+-+%5Cmu%29%5E2%5Cright%5D&bg=ffffff&fg=000&s=0&c=20201002)
![E\left[\sum(x_i - \bar{x})^2\right] = \sum E[(x_i - \mu)^2] - N \cdot E[(\bar{x} - \mu)^2]](https://s0.wp.com/latex.php?latex=E%5Cleft%5B%5Csum%28x_i+-+%5Cbar%7Bx%7D%29%5E2%5Cright%5D+%3D+%5Csum+E%5B%28x_i+-+%5Cmu%29%5E2%5D+-+N+%5Ccdot+E%5B%28%5Cbar%7Bx%7D+-+%5Cmu%29%5E2%5D&bg=ffffff&fg=000&s=0&c=20201002)



![\text{Cov}(X, Y) := \mathbb{E}[(X - \mu_X)(Y - \mu_Y)]](https://s0.wp.com/latex.php?latex=%5Ctext%7BCov%7D%28X%2C+Y%29+%3A%3D+%5Cmathbb%7BE%7D%5B%28X+-+%5Cmu_X%29%28Y+-+%5Cmu_Y%29%5D&bg=ffffff&fg=000&s=0&c=20201002)
Covariance Matrix!!
Multidimensional Data
For a 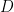
![\mathbf{X} = [X_1, \dots, X_D]^\top](https://s0.wp.com/latex.php?latex=%5Cmathbf%7BX%7D+%3D+%5BX_1%2C+%5Cdots%2C+X_D%5D%5E%5Ctop&bg=ffffff&fg=000&s=0&c=20201002)
![\boldsymbol{\mu} = \mathbb{E}[\mathbf{X}]](https://s0.wp.com/latex.php?latex=%5Cboldsymbol%7B%5Cmu%7D+%3D+%5Cmathbb%7BE%7D%5B%5Cmathbf%7BX%7D%5D&bg=ffffff&fg=000&s=0&c=20201002)

![\mathbf{\Sigma} := \mathbb{E}[(\mathbf{X} - \boldsymbol{\mu})(\mathbf{X} - \boldsymbol{\mu})^\top]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7B%5CSigma%7D+%3A%3D+%5Cmathbb%7BE%7D%5B%28%5Cmathbf%7BX%7D+-+%5Cboldsymbol%7B%5Cmu%7D%29%28%5Cmathbf%7BX%7D+-+%5Cboldsymbol%7B%5Cmu%7D%29%5E%5Ctop%5D&bg=ffffff&fg=000&s=0&c=20201002)
The entry at position 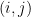
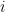


The diagonal elements are the variances of each component, 
As always, properties!
1. Symmetry: 

2. Positive Semi-Definiteness: For any vector 


Empirical Covariance Matrix
As always, we only have a sample; not the distribution / population.
So, what do we do?
I will first define population and samples since I’ve just been naming them
- Population: The entire set of all possible observations of interest. Population parameters, such as the true mean
, are considered fixed but unknown constants.
- Sample: A finite subset of observations $latex {\mathbf{x}n}{n=1}^N$ drawn from the population. We use the sample to make inferences about the unknown population parameters.
With me so far? Good, now more definitions.
An estimator is a function of the sample data that is used to estimate an unknown population parameter. For example, the sample mean
is an estimator for the population mean
. A crucial property of an estimator is its bias.
Definition (Bias of an Estimator): Let
be an unknown population parameter and let
be an estimator of
![\text{Bias}(\hat{\theta}) = \mathbb{E}[\hat{\theta}] - \theta](https://s0.wp.com/latex.php?latex=%5Ctext%7BBias%7D%28%5Chat%7B%5Ctheta%7D%29+%3D+%5Cmathbb%7BE%7D%5B%5Chat%7B%5Ctheta%7D%5D+-+%5Ctheta&bg=ffffff&fg=000&s=0&c=20201002)
- An estimator is unbiased if its bias is zero, i.e.,
. This means that if we were to repeatedly draw samples of size
- An estimator is biased if
.
(See? I told you a few more sections earlier; keyword: biased-estimator-def)
Cool, I just want to remind you of Bessel’s Correction once again:
![\mathbb{E}[\hat{\sigma}^2_{N-1}] = \mathbb{E}\left[\frac{N}{N-1}\hat{\sigma}^2_N\right] = \frac{N}{N-1}\mathbb{E}[\hat{\sigma}^2_N] = \frac{N}{N-1} \left( \frac{N-1}{N}\sigma^2 \right) = \sigma^2](https://s0.wp.com/latex.php?latex=%5Cmathbb%7BE%7D%5B%5Chat%7B%5Csigma%7D%5E2_%7BN-1%7D%5D+%3D+%5Cmathbb%7BE%7D%5Cleft%5B%5Cfrac%7BN%7D%7BN-1%7D%5Chat%7B%5Csigma%7D%5E2_N%5Cright%5D+%3D+%5Cfrac%7BN%7D%7BN-1%7D%5Cmathbb%7BE%7D%5B%5Chat%7B%5Csigma%7D%5E2_N%5D+%3D+%5Cfrac%7BN%7D%7BN-1%7D+%5Cleft%28+%5Cfrac%7BN-1%7D%7BN%7D%5Csigma%5E2+%5Cright%29+%3D+%5Csigma%5E2&bg=ffffff&fg=000&s=0&c=20201002)
and also the why once again – The bias arises because we are measuring deviations from the sample mean
The data points are, on average, closer to their own sample mean than to the true population mean, leading to an underestimation of the true spread.
First, the MLE!
Maximum Likelihood Estimation is a fundamental method for deriving estimators. Given a dataset
and a parameterized probability model
, the likelihood function is the probability of observing the data, viewed as a function of the parameters
:
The MLE principle states that we should choose the parameters
that maximize this likelihood function. For an i.i.d. sample, this is equivalent to maximizing the log-likelihood:
How did we get the above? Take log on both sides of
Ok what the hell is this? Probability theory solves the “forward problem”: Given a model with known parameters, what does the data look like? (e.g., “If this coin is fair,
, I expect to see roughly 5 heads in 10 flips.”)
(Statistical inference in general, and) MLE in particular, solves the inverse problem: Given the data we have observed, what were the most likely parameters of the model that generated it? (e.g., “I flipped a coin 10 times and got 8 heads. What is the most plausible value for this coin’s bias, $\theta$?”)



- Normally, we think of
as a probability distribution over possible datasets
, for a fixed parameter setting
- The likelihood function,
, treats the observed data
Here, I present a statement:
The covariance estimator with denominator
Ponder this. What can it even mean? Yeah, it’s just that
Specifically, we assume our data points 


That means that the log-likelihood function for this model and a dataset

Do math, take partial derivatives, yada yada and we get:
(the sample mean)

Takeaway – The MLE (estimator / estimation – same thing) is the estimator that makes the observed data “most probable” under a Gaussian model.
Now, this estimator is biased!
Changing notation a bit (I started this on another day and am lazy to keep track, haha).
Here’s what’s up rn:
The estimator for the covariance matrix using denominator

Its expected value is: (tiny recap time!)
![\mathbb{E}[\mathbf{S}_N] = \frac{N-1}{N}\mathbf{\Sigma}](https://s0.wp.com/latex.php?latex=%5Cmathbb%7BE%7D%5B%5Cmathbf%7BS%7D_N%5D+%3D+%5Cfrac%7BN-1%7D%7BN%7D%5Cmathbf%7B%5CSigma%7D&bg=ffffff&fg=000&s=0&c=20201002)
This estimator is biased. The unbiased sample covariance matrix is:

Its expected value is ![\mathbb{E}[\mathbf{S}_{N-1}] = \mathbf{\Sigma}](https://s0.wp.com/latex.php?latex=%5Cmathbb%7BE%7D%5B%5Cmathbf%7BS%7D_%7BN-1%7D%5D+%3D+%5Cmathbf%7B%5CSigma%7D&bg=ffffff&fg=000&s=0&c=20201002)
Now, which one do we use?
Use MLE for ML, unbiased for statistics.
- Statistics: Unbiasedness is a highly desirable property. An unbiased estimator is correct on average.
- The unbiased estimator
is the standard. In statistical analysis (e.g., hypothesis testing, confidence intervals), using an unbiased estimate of variance is critical for the validity of the statistical procedures. The focus is on estimation accuracy.
- The unbiased estimator
- Machine Learning: Minimizing prediction error is a desired property. MLE is better for such cases.
- The quality of an estimator is often judged by its Mean Squared Error (MSE)
- This is the bias-variance tradeoff. The best estimator is one that minimizes this total error.
- The MLE estimator
has a slightly lower variance than the unbiased estimator
. Therefore, if the goal is to find an estimator that is “closer” to the true value on average in terms of squared error, the biased MLE is superior.
![\text{MSE}(\hat{\theta}) = \mathbb{E}[(\hat{\theta} - \theta)^2] = (\text{Bias}(\hat{\theta}))^2 + \text{Var}(\hat{\theta})](https://s0.wp.com/latex.php?latex=%5Ctext%7BMSE%7D%28%5Chat%7B%5Ctheta%7D%29+%3D+%5Cmathbb%7BE%7D%5B%28%5Chat%7B%5Ctheta%7D+-+%5Ctheta%29%5E2%5D+%3D+%28%5Ctext%7BBias%7D%28%5Chat%7B%5Ctheta%7D%29%29%5E2+%2B+%5Ctext%7BVar%7D%28%5Chat%7B%5Ctheta%7D%29&bg=ffffff&fg=000&s=0&c=20201002)
Back to PCA
We take the empirical covariance matrix to be

where 

Notice that the covariance matrix, 
Important Note: Ok, cool,
(Perspective 1) As maximising variance
Remember: We have centered the dataset!
Now, we are trying to find a lower dimension space. So, we reduce the number of dimensions (duh), but what this means is that we should be able to reflect 
So, we try to find a set of coordinate vectors 

Now, let us find the direction, represented by a unit vector 
The projection of a data point 







What we want to do is maximize this variance subject to the constraint that 

We form the Lagrangian:

Setting the gradient with respect to

This is the eigenvalue equation for the covariance matrix 


Subsequent principal components
are found by finding the eigenvectors of
Notice how it is also very neat that the variance is then just the sum of the chosen eigen values:)
(Perspective 2) As minimizing reconstruction error
No one really talks about this one much but let’s see. I will be very math-y here.
- Let the principal subspace be an
-dimensional subspace spanned by an orthonormal basis of vectors
.
- The orthogonal projection of a data point
.
- Our goal is to find the basis
that minimizes the average squared reconstruction error:


By the properties of orthogonal projections, the total variance can be decomposed: 


So, yes, we have very smartly reduced the 2nd perspective to the first perspective. Yay!
Probabilistic PCA (PPCA)
Classical PCA is a geometric, algorithmic procedure. Probabilistic PCA (PPCA) reformulates this procedure within a probabilistic framework by defining a latent variable model that describes the generative process of the data.
What? Ok, fine, I’ll explain.
PCA is a point estimate – gives you a single, definitive answer for the principal components and the lower-dimensional space they define, without expressing any uncertainty about that result. It is fundamentally a descriptive tool, not a generative one. This means:
- It cannot generate new data points
- It does not provide a likelihood of the data, making principled model selection (e.g., choosing the optimal number of components
- It cannot handle missing values in the data.
So, we now have PPCA! PPCA posits that the high-dimensional observed data 


PPCA models the relationship between the latent and the observed (this is such a smart sounding statement, omg).
Definitions
(No, what did you expect?)
Latent Variable Prior: A simple cause is assumed for each data point. This is modeled by a prior distribution over the latent variable 

This asserts that the latent variables are, a priori, independent and centered at the origin of the latent space.
Conditional Distribution of Data (Likelihood): The observed data point 


This is equivalent to the equation 
is a matrix that maps the latent space to the data space. Its columns can be interpreted as the basis vectors of the principal subspace.
- The principal subspace is the lower-dimensional plane or hyperplane that captures the most important variations in your data. Basis vectors are the fundamental directions that define or “span” that subspace.
- This just means that the reconstructed data point is made by a linear combination of the basis vectors of the principal subspace (plus the following things).
is the mean of the data.
- Yeah I don’t think I can explain this more.
is isotropic Gaussian noise, which accounts for the variability of data off the principal subspace.
- isotropic = fluctuations are same in all directions; hence the scalar multiplied with the identity matrix for variance.
- Also, every dimension is independent and uncorrrelated.
Calculation time!
To define the likelihood of the model, we must integrate out the latent variables to find the marginal distribution of the observed data, 

Since both 

- Mean:
.
- Covariance:
.
The parameters of the model (
Advantanges
- Handling Missing Data: The generative nature of PPCA allows it to handle missing data through the Expectation-Maximization (EM) algorithm.
- How? Again, I think I’ll write more on EM later. But, for now, let’s take it as is.
- Model Selection: The probabilistic formulation provides a log-likelihood value for the dataset given a model.
- This allows for principled model selection, for instance, choosing the optimal number of principal components $M$ by comparing the likelihood values (often using criteria like AIC or BIC) for different choices of $M$.
- PPCA is used in robotics for sensor fusion and state estimation where sensor noise is a critical factor. How? Idk – I took this statement as is. Takeaway – super useful.
Kernel PCA
This addresses the primary limitation of standard PCA – PCA seeks a linear subspace to represent the data. If the data lies on a complex, non-linear manifold (e.g., a spiral, a “Swiss roll”), any linear projection will fail to capture the underlying structure, leading to a poor low-dimensional representation.
How? Map the data from the original input space 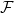

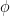

But, we get a computational barrier:
- The dimensionality of
- The function
So, we use the kernel trick!
Let’s perform PCA on the mapped data $latex {\phi(\mathbf{x}n)}{n=1}^N$. The covariance matrix in the feature space

The eigenvalue problem in 

Want to go on? Follow this lecture pdf from slide 32; I really can’t write it better for myself either. The only extra thing that I can offer is to answer the question: Why can eigenvectors be written as linear combiunation of features?
The eigenvectors vⱼ can be written as a linear combination of the features because they must lie in the space spanned by the feature vectors φ(xᵢ) themselves.
Let’s start with the PCA equation:

We can isolate the eigenvector vⱼ by rearranging the equation (assuming the eigenvalue λⱼ is not zero):

Now, let’s group the terms differently:

Notice that the term in the parenthesis, 
If we call this scalar coefficient 

This proves that any eigenvector vⱼ is simply a weighted sum of all the feature vectors φ(xᵢ).
Spectral Clustering
This is a graph-based clustering technique that partitions data by analyzing the spectrum (eigenvalues) of a graph’s Laplacian matrix.
Foundations
What? Ok, some definitions :sparkles: (I hope you can relate to some things if you took data structures and algorithms)
Similarity Graph
Given a dataset of 

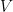
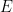


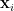

- Common ways to construct
include the k-nearest neighbor graph or using a Gaussian kernel:
.
Graph Partitioning
The goal of clustering is to partition the vertices 
- This is our objective btw in case it wasn’t clear.
Graph Laplacian
Ok, this isn’t much of a definition. I’ll try to explain it since it’s the core component / concept.
It is the fundamental object whose spectrum (eigenvalues and eigenvectors) reveals the cluster structure of the graph.
First, let’s define the degree matrix. A diagonal matrix where each diagonal element 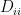

The unnormalized graph laplacian is:

Some properties of the Graph Laplacian:
- It is symmetric and positive semi-definite.
- It is symmetric since it’s the combination of the adjacency matrix and the degree matrix (a diagonal matrix) are symmetric.
- We later show that
turns into a square term, so it is positive semi-definite (or semi positive definite). Or, ctrl + F and find quadratic-laplacian-square.
- The Spectral Theorem states that any real, symmetric matrix has real eigenvalues and orthogonal eigenvectors.
- The added property of being positive semi-definite ensures these real eigenvalues are also non-negative (λ≥0).
- This is crucial because it allows us to reliably sort the eigenvalues from smallest to largest.
- The smallest eigenvalue of
is always
, with the corresponding eigenvector being the vector of all ones,
.
- This is because the sum of each row in the Laplacian matrix is always zero by construction (
).
- This is because the sum of each row in the Laplacian matrix is always zero by construction (
Graph cut and Laplacian
Ok, simplest case first – we have two partitions, i.e., two disjoint sets, 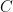

The “cut” is the sum of weights of all edges connecting these two sets:

Repeating for dramatic effect – minimizing this cut directly is an NP-hard combinatorial problem.
We (I) define a partition indicator vector 
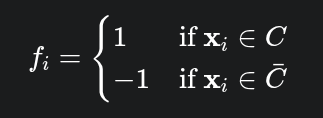
Yeah, I didn’t want to write latex.
Now, consider the quadratic form of the Laplacian, 






See? I promised you a square earlier! (keyword – quadratic-laplacian-square)
Now, observe the term 
- If
(i.e., both nodes are in the same partition),
.
- If
(i.e., nodes are in different partitions),
.
So / Therefore / Thus / Hence, minimizing the graph cut is equivalent to minimizing
How does this help us?
The optimization problem 

So, what we do is – we relax. With multiple xs. The entries of 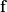

This is the formulation for minimizing the Rayleigh quotient. Its solution? Notice how its the same optimization problem we had in PCA?
The solution is given by the eigenvector of
However, the smallest eigenvalue is 
So, the non-trivial solution must be orthogonal to this vector of ones.



This is the eigenvector corresponding to the second-smallest eigenvalue of
- This eigenvector is known as the Fiedler vector.
Generalized
(I’m just going to get an LLM to generate it)
- Construct Similarity Graph: Given
- Compute Laplacian: Compute the degree matrix
and the Laplacian
.
- Compute Eigenvectors: Find the first
eigenvectors
of
- Form Embedding: Construct a matrix
with these eigenvectors as its columns.
- Cluster Embedding: Treat each row of
as a new data point in a
- Assign Original Points: The cluster assignment of the
t-Distributed Stochastic Neighbor Embedding (t-SNE)
Why not just PCA?
Remember kernel PCA? The one that solves the problem of PCA being linear? Yeah, t-SNE solves the same thing.
Prerequisites
Gaussians
The bell curve, duh. I am not explaining this further. Here’s the formula:

The important thing to know here is that Gaussians have light tails.
- Values away from the mean are exceptionally unlikely.
t-distribution
These have heavier tails than Gaussians – defined by degrees of freedom (
- The PDF of the t-distribution decays polynomially, not exponentially.
- As
, the t-distribution converges to the Gaussian distribution.
- The specific case used in t-SNE is the t-distribution with one degree of freedom (

Now, before moving on, I want to add a bit more context – the teacher is the gaussian and the student is the t-distribution (the lower dimension one).
This is a crucial design choice to solve the “crowding problem”. In a 2D map, there isn’t enough space to accurately represent all the neighborhood distances from a high-D space. The heavy tails of the t-distribution mean that two points can be placed far apart in the map and still have a non-trivial similarity score. This allows t-SNE to place moderately dissimilar points far apart, effectively creating more space for the local neighborhoods to be modeled without being “crowded” together.
Kullback-Leibler (KL) Divergence
It is a measure of how one probability distribution,
, is different from a second, reference probability distribution,
. It is not a true distance metric (it is not symmetric), but it is often used to measure the “distance” or “divergence” between distributions.
For discrete distributions over outcomes
Again, context – this is our cost function in t-SNE.

Oh? You want more on it? I want to cover both KL divergence and Gaussians more. But, for now, I think I can suggest Wikipedia.
Math
Three stages:
Stage 1: Modeling High-Dimensional Similarities
The similarity between two data points, 

So, sorta like “j given i” but not exactly.
Ok, what is 
Perplexity
(adding this as well – notice how it dictates the size of the neighbourhood in a way)
Yeah, you see how? (I just added the last sentence within brackets for fun)
Now, notice how the scale parameter 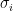
Before everything else, focus on the following definition.
For each data point 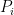


The entropy 
- If the distribution is sharply peaked on one neighbor (low uncertainty), the entropy will be low.
- If the distribution is spread out over many neighbors (high uncertainty), the entropy will be high.
The perplexity of the distribution

(If you’ve done NLP, this would seem very familiar. That’s because it is.)
What does this do? Notice that if the probability distribution $P_i$ is uniform over $k$ neighbors (and zero for all others), its perplexity will be exactly $k$.
Perplexity can be interpreted as a smooth, continuous measure of the effective number of neighbors for point
Ok, now, we don’t know
Yes. So, here’s what we do:
- User Input: The user specifies a single, global perplexity value. This value is the same for all points and typically ranges from 5 to 50. Let’s say the user chooses a perplexity of 30.
- Per-Point Optimization: For each data point
- The Search Algorithm: This search is performed efficiently using binary search. The algorithm makes a guess for
Neat, isn’t it?
Last thing in this stage – Symmetrization.
The conditional probabilities

where
Stage 2: Modeling Low-Dimensional Similarities (
Now, we define a similar probability distribution 
Remember how I told you about the crowding problem? No? Go back.
The t-distribution is heavy-tailed, meaning it decays much more slowly with distance than a Gaussian.

This is the student’s t-distribution with one degree of freedom.
Stage 3: Optimization via KL Divergence Minimization
We minimize the KL divergence:

If you are a dum-dum like me – 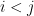
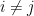
This is non-convex: has multiple minimas. So, we use iterative gradient descent and hope for the best.

(This makes t-SNE non-deterministic!)
Why not just kernel PCA?
Kernel PCA: finds an optimal linear projection in a non-linearly transformed space.
- Non-linear feature extraction and dimensionality reduction. It seeks to find a set of non-linear components that can be used for subsequent tasks (e.g., classification, regression).
t-SNE: finds a a faithful representation of the local data topology.
- Visualization and exploratory data analysis. It seeks to create a 2D or 3D map of the data that reveals its underlying local structure and clustering.
Now, a very, very important point is that
Kernel PCA deals with global structure, t-SNE with local.
You use kernel PCA when you need non-linear feature extraction for a subsequent machine learning task.
You use t-SNE for visualizing / exploring a high-dimensional feature space.
Key takeaway: Do NOT use t-SNE for feature extraction.

Leave a reply to Matrix Decompositions 3 – Vineeth Bhat Cancel reply